-
Ground-state Structural Disorder and Excited-state Symmetry Breaking in a Quadrupolar Molecule
M. Soederberg, B. Dereka, A. Marrocchi, B. Carlotti and E. Vauthey
Journal of Physical Chemistry Letters, 10 (2019), p2944-2948


DOI:10.1021/acs.jpclett.9b01024 | unige:126821 | Abstract | Article HTML | Article PDF | Supporting Info
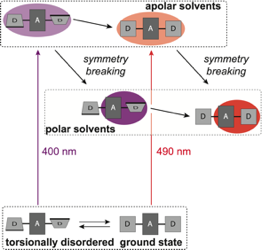
The influence of torsional disorder around the ethynyl pi-bridges of a linear D-pi-A-pi-D molecule on the nature of its S1 excited state was investigated using ultrafast time-resolved infrared spectroscopy. By tuning the pump wavelength throughout the S1<- S0 absorption band, subpopulations with different extents of asymmetry could be excited. In non-polar solvents, the equilibrated S1 state is symmetric and quadrupolar independently of the initial degree of distortion. Photoexcitation of distorted molecules is followed by planarization and symmetrization of the S1 state. Excited-state symmetry breaking is only observed in polar environments, where the equilibrated S1 state has a strong dipolar character. However, neither the extent nor the rate of symmetry breaking are enhanced in an initially distorted molecule. They are only determined by the polarity and the dynamic properties of the solvent.